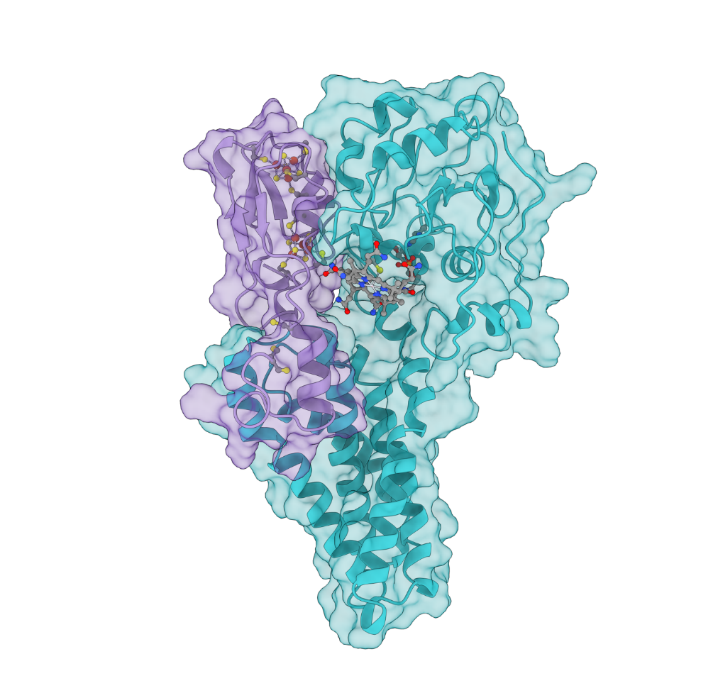
A team led by the Department of Energy’s Oak Ridge National Laboratory created a computational model of the proteins responsible for the transformation of mercury to toxic methylmercury, marking a step forward in understanding how the reaction occurs and how mercury cycles through the environment.
Methylmercury is a potent neurotoxin that is produced in natural environments when inorganic mercury is converted by microorganisms into the more toxic, organic form. In 2013, ORNL scientists announced a landmark discovery: They identified a pair of genes, hgcA and hgcB, that are responsible for mercury methylation.
Those genes encode the proteins HgcA and HgcB, whose structure and function ORNL scientists have been working to better understand.
“Determining protein structures can be challenging,” said Jerry Parks, the head investigator and leader of the Molecular Biophysics group at ORNL.
These two proteins are difficult to characterize experimentally for several reasons: they are produced by anaerobic microorganisms and are therefore highly sensitive to oxygen; they are expressed at such low levels in cells that they are barely detectable by conventional techniques; HgcA is embedded in the membrane of a cell, making it more challenging to study than a soluble protein; and both proteins have complex cofactors—substances that bind to proteins and are essential for their function.
“We don’t have an experimental structure yet for these proteins, so the next best thing is to use computational techniques to predict their structure,” Parks said.
The computational model was generated using a large dataset of HgcA and HgcB protein sequences from many different microorganisms, ORNL’s high-performance computing resources and bioinformatics, and structural modeling techniques as detailed in a recent article in Communications Biology.
The result is a 3D structural model of the HgcAB protein complex and its cofactors that scientists can use to develop new hypotheses designed to understand the biochemical mechanism of mercury methylation and then test them experimentally.
Scientists have been predicting protein structures from their amino acid sequences for many years. In 2017, a team led by the University of Washington reached a milestone, modeling the structures of hundreds of previously unsolved protein families by mining large metagenomic datasets for diverse protein sequences. This approach predicts which pairs of amino acids in each protein are in close contact with each other, and then uses that information to fold the proteins computationally.
Parks was eager to apply the same techniques to the mercury work, turning to data available from DOE’s Joint Genome Institute, a DOE Office of Science user facility.
The scientists searched the JGI database for HgcA and HgcB amino acid sequences. They then performed a coevolution analysis to identify coordinated changes that occur among pairs of amino acids. Coevolution makes it likely that those coordinated pairs are close to each other in the three-dimensional folded structure of the protein. This information can be used to guide computational protein folding and predict how the folded protein domains interact with each other.
One surprising finding by the team is that the two domains of HgcA don’t interact with each other, but they both interact with the HgcB protein. The model also suggests that conserved cysteine amino acids in HgcB are likely involved in shuttling some forms of mercury, methylmercury, or both, to HgcA during the reaction. Some features of these proteins are similar to other more well-studied proteins, but others are unique and have not been observed before in any other protein.
Future research will involve experimental testing. Stephen Ragsdale’s group at the University of Michigan is working out a way to produce the HgcA and HgcB proteins in E. coli bacteria in sufficient quantities to enable the proteins to be studied in the laboratory using spectroscopic techniques and X-ray crystallography. “We are excited that this important experimental work is being done,” said Parks. “It will be interesting to see how well we did with our structure predictions.”
Mercury is a naturally occurring element found worldwide, and scientists at ORNL have come to realize that the microorganisms that convert inorganic mercury to methylmercury are also widespread. “We don’t know as much as we’d like about all the different reactions and processes that mercury can undergo,” Parks added. “This work helps us understand more about one of the most important biotransformations of mercury in nature.”
In addition to gaining insight into mercury methylation, the project creates a new capability at ORNL that can be used to explore the structure and function of other microbial proteins. In particular, Parks and colleagues are interested in characterizing proteins from microorganisms referred to as microbial dark matter because they are unable to be cultured in the lab and are otherwise difficult to study.
“There is so much we still don’t know about all the unusual proteins that are produced by microorganisms,” Parks said. “This technique allows us to begin characterizing these complex, mysterious biological systems.”
Other scientists working on the project at ORNL include Connor Cooper, Alexander Johs, Brian Sanders and Mircea Podar. Researchers at other institutions were Sergey Ovchinnikov from Harvard University; Kaiyuan Zheng, Katherine Rush and Stephen Ragsdale at the University of Michigan; and Georgios Pavlopoulos and Nikos Kyrpides from the JGI at Lawrence Berkeley National Laboratory.
The research was funded by the DOE Office of Science and the Laboratory Directed Research and Development program at ORNL.
ORNL is managed by UT-Battelle for the U.S. Department of Energy's Office of Science, the largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, please visit https://energy.gov/science.



